
A short introduction and overview of the requirements
In 2017, the European Commission released the new Medical Device Regulation (MDR) that will replace the Medical Device Directive (MDD) in May 2021. The regulation has been written to assure better quality and improve the safety aspects for all medical devices that are sold on the European market.
The biggest changes include a more extensive vigilance system that needs to ensure that (serious) incidents with a medical device are identified, reported and trended. In case of an incident, related safety corrective actions are needed, like a recall or safety notice to healthcare professionals. The manufacturer’s vigilance system should initiate the action and inform competent authorities.
Besides the more extensive vigilance system, the manufacturer will have to implement an overarching post-market surveillance (PMS) system that actively collects various types of product related information throughout the entire lifetime of the device. The PMS is both reactive and proactive. Vigilance is part of the reactive PMS, while a post-market clinical follow-up study is part of the proactive PMS.
This information should be periodically analysed to evaluate the quality, performance and safety of the product after it has been released on the market. With the evaluation results, the necessary actions are to be taken to improve quality, performance or safety aspects of the device. These necessary actions can go from detecting trends to updating the risk-benefit analysis and eventually creating a CAPA or even redesigning the medical device.
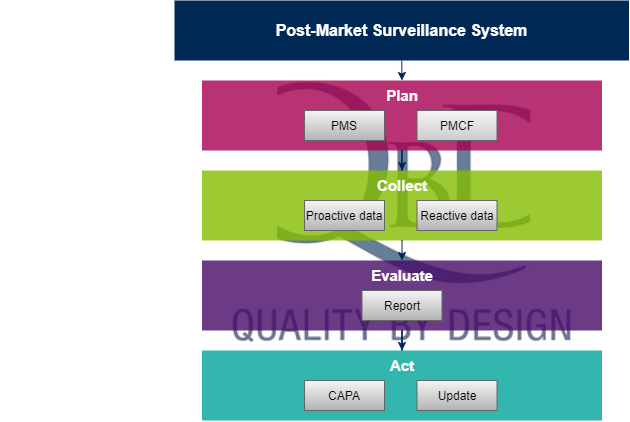
PMS plan
Once you have your PMS system set-up with all the necessary procedures, instructions and templates to be able to actively gather and analyse post-market data, it’s time to write a PMS plan for every medical device that you sell on the European market. The PMS plan becomes part of the technical documentation of the medical device (Medical Device File or Technical File) and describes which information is gathered (input data) to evaluate the quality, performance and the safety of the device. Examples of data that should be actively gathered are customer complaints, production data and literature (regarding your devices or devices that are similar). The plan should also mention how and at what frequency the data will be collected. Other PMS information comes from maintenance reports and product related corrective actions, but also marketing and sales figures.
PMS report (Class I)
In case of a risk class I device, a PMS report should be created. It should contain a summary of the information on the device that is collected and evaluated. It should also point out the actions that will be taken. In the MDR, nothing is said about the frequency with which this report needs to be updated, but it needs to be available for Notified Body and Competent Authority. When it comes to a newly developed product, we advise to create a PMS report at least yearly. If after a few years the data do not show concerns, the frequency could be decreased.
PSUR (Class IIa/IIb/III)
For higher risk classes, a Periodic Safety Update Report (PSUR) is required. This report is more extensive compared to a PMS report. It should include a rationale and a description of all the corrective and/or preventive actions taken based on the analysed product information. During the lifetime of a medical device, the PSUR must also set out:
- the conclusions of the benefit-risk determination of the device
- the main findings of the PMCF
- the volume of sales
- an evaluation of the population that uses the device
When the risk class of the device is IIa, the PSUR should be updated at least every two years, while you should update it at least annually if the risk class is IIb or III. In case the device is an implantable or belongs to risk class III, this report should also be uploaded to the EUDAMED database, where your Notified Body can consult it. The Notified Body will then write an evaluation and upload it to the same electronic system, where the report and evaluation are also made available for the Competent Authority. PSUR of Class IIa devices do not have to be uploaded to EUDAMED for an evaluation, but should be made available for the Notified Body and Competent Authority.
PMCF
Besides product-related technical data, it’s also important to keep collecting clinical data of the device, the so called Post Market Clinical Follow-up (PMCF). PMCF is part of the device’s PMS plan and is especially needed when the device:
- contains novel innovative medical technology
- presents a high risk during use
- is used at high-risk anatomical locations
- is used for high-risk populations (e.g. children, pregnant women, elderly)
But also when few clinical data about long-term behaviour of the device is available. When a PMCF plan is not created, a (good) rationale should be included in the PMS plan. A PMCF plan needs to be written to make sure the clinical information is gathered, evaluated and updated throughout the whole life cycle of the device. The plan includes the methods and procedures that will be used, a rationale for the appropriateness of those methods, evaluation of clinical data from equivalent devices and a time schedule.
Findings from the PMCF shall be documented in a PMCF evaluation report, which will be part of the technical documentation of the device. The conclusions of this report could lead to an update of the overall clinical evaluation of the device. The most important new findings of the PMCF studies need to be introduced in the PSUR as well. Just like with the PMS and PSUR, any corrective and/or preventive actions must be identified and implemented based on the evaluation results.