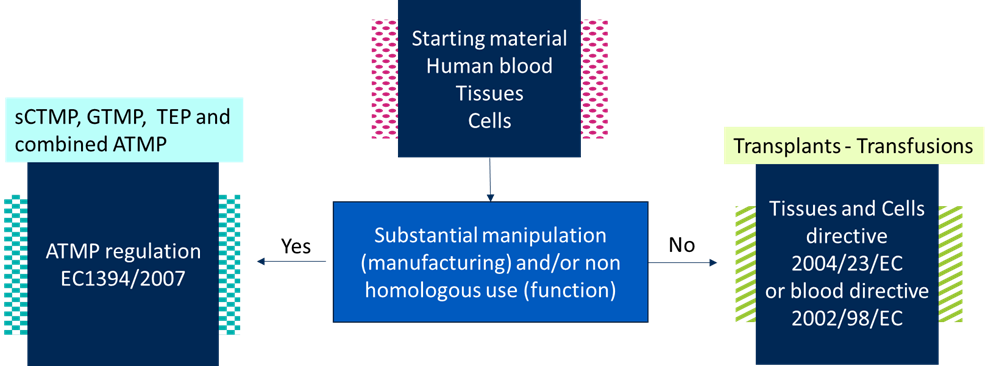
Do you want to distinguish starting materials used for ATMPs from those used for transplants or transfusions? This distinction is made based on how the starting materials are processed and/or used clinically. We’ve listed the steps to make such a distinction, with the help of a flowchart.
Current ATMP workflows start with donor selection and procurement of autologous (i.e. from patient), allogeneic (i.e. from human donor) or xenogeneic (i.e. from animal) starting material. These starting materials include (human) blood, tissues and cells.
The procurement, handling and patient/donor specific differences of starting materials have a significant impact on the manufacturing process. Therefore, raw material variability is an important input factor that needs to be considered during product development and manufacturing. A thorough understanding of the critical material attributes (CMA) and critical process parameters (CPP) in correlation with their effect on critical quality attributes (CQA) is highly recommended to develop a robust platform.
Additionally, the raw materials (defined as ancillary materials in USP <1043>) used during manufacturing and storage will significantly influence the characterisation and processability of an ATMP. Materials are defined as raw (ancillary) materials if they are used in the manufacturing process of gene, cell or tissue-engineered products but are not intended to be part of the final product. For example: fetal bovine serum, digestion enzymes (e.g. collagenase, DNAse), growth factors, cytokines, monoclonal antibodies, antibiotics, resins, cell-separation devices and media components.1
Selecting the appropriate starting and raw materials is a crucial step to prevent delays, repetition of work or to avoid unforeseen costs during upscaling. Furthermore, the choice can depend on regulations or requirements set by regulators and the industry.
How to select the correct raw materials?
The selection of raw materials needs to be based on a thorough screening of the vendor, characterisation of the material and the proven suitability of the material for the intended manufacturing process. The supply chain of the raw materials needs to be secure and reliable. To ensure this, a qualification of the supplier must be performed, and quality agreement contracts must be formulated. Designated documents to consult for selecting the raw materials and start up your development project are:
- EudraLex volume 4, part IV chapter 7 starting and raw materials
- General chapter on raw materials of biological origin for the production of cell based and gene therapy medicinal products (EP 5.2.12)
- USP <1046> Cell and gene therapy products
- USP <1047> Gene Therapy Products
- USP <1043> Ancillary materials for cell, gene and tissue-engineered products. Obligatory lecture
In addition, we highlight some specific topics to guide you through the decision-making process:
- Raw material source
Is your raw material of human or animal blood origin, produced using substances of human or original origin, or free from substances of human or animal origin?
It is important to minimize the risk of transmitting adventitious agents. Since human blood and tissue derived starting materials are used in the production of ATMPs, the tracing and testing (of donors) for infectious transmissible agents is a key element (in line with the directive 2004/23/EC and 2002/98/EC). Viral safety aspects, as specified in the general text of the EP 5.1.7, also need to be considered and respected.
More information can be found in the note for guidance on minimizing the risk of transmitting animal spongiform encephalopathy agents via human and veterinary medicinal products (EMA/410/01 rev.3), CHMP/CAT position statement on Creutzfeldt-Jakob disease and advanced therapy medicinal products (EMA/CHMP/BWP/353632/2010) and WHO Guidelines on Tissue Infectivity Distribution in Transmissible Spongiform Encephalopathies. - Production process
Raw materials need to have testing certificates for sterility or known microbial contamination, endotoxin level, impurities and other predefined quality requirements.
Because cell therapies cannot be terminally sterilized by membrane filtration, the production steps are performed in an LAF grade A with B background or isolator with C or D background. In some conditions, other clean area grades can be allowed, which are specified in chapter 9.5 of EudraLex volume 4, part IV.2 Due to this sterilization limitation stringent sourcing requirements and acceptance requirements are applicable. - Quality requirements
Quality requirements for raw materials need to be pre-defined for identity, purity and biological activity.
For example, the osmolarity (EP 2.2.35), pH (EP 2.2.3), mycoplasma (EP 2.6.7), endotoxins (EP 2.6.14) needs to be measured and tested to fulfill the necessary requirements. Regulators admit that raw materials are often only available with research grade claim2 and their quality, safety and consistency is difficult to assess. Sera from humans or animal sources are typical examples of complex biological mixtures whose composition cannot always be fully determined or defined. Manufacturers should be aware of these risks and should provide details on the suitability and possible use of the material. These details should include/focus on the identity, safety and functionality of the material. However, this combined information is often lacking 2.
The process of selecting the appropriate raw materials and at the same time considering all current guidelines can be quite challenging. The experienced consultants of QbD can help you meet these ATMP regulatory requirements. Our cell by design format is able to evaluate the impact of raw material variability, identify the critical process parameters and define the critical quality attributes of the product. Feel free to contact us for more information.
1EMA/CAT/852602/2018 Draft guideline on quality, non-clinical and clinical requirements for investigational advanced therapy medicinal products in clinical trials.
2Part IV of EudraLex volume 4 Guidelines on Good Manufacturing Practice specific to Advanced
Therapy Medicinal Products.